This repository implements a deep learning-guided virtual screening protocol for identifying potential inhibitors of the NirA protein, using an ultra-large chemical library. The workflow is based on the original Deep Docking framework by James Gleave, modified and optimized for phytochemical screening against NirA, a key enzyme of interest in this study.
The methodology combines deep neural network-based filtering with molecular docking to rapidly prioritize promising binders from a massive compound library.
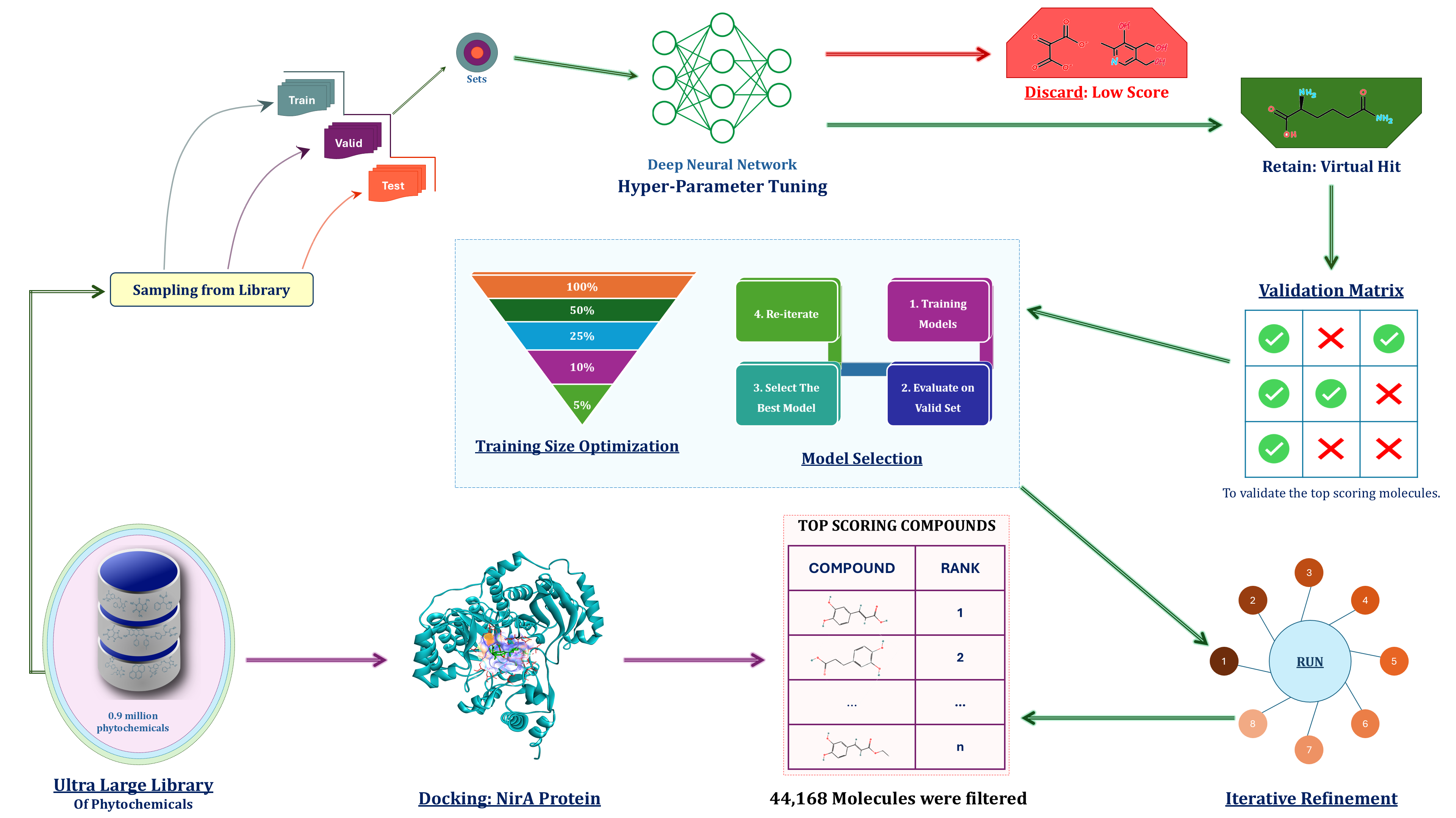
- Sampling from Library: Compounds are sampled from public databases (ZINC, DUDE, IMPPAT, COCONUT).
- Data Preparation: Decoys and actives are prepared based on molecular weight and activity criteria.
- Dataset Split: ~9 million phytochemicals are split into:
- Training
- Validation
- Testing
- Deep Neural Network Training:
- Hyperparameter tuning
- Training set size optimization
- Model selection
- Virtual Screening:
- Predict docking scores
- Discard low-score compounds
- Retain top hits as virtual hits
- Validation: Top hits are validated using molecular docking against the NirA protein.
- Iterative Refinement: Top compounds are re-scored, re-trained, and filtered in successive rounds.
-
Library Size: ~9 million
-
Top Scoring Hits: 44,168 compounds
-
Databases: ZINC, DUDE, IMPPAT, COCONUT
-
Architecture: Feed-forward Deep Neural Network with hyperparameter tuning and iterative refinement
If you use this repository or base your work on it, please cite:
-
Ton et al. (2020) Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery DOI
For queries, contact: thedrsoham[at]gmail[dot]com or open an issue in this repository.
"Accelerating ultra-large-scale docking with deep learning for NirA-targeted compound discovery."