
![]()
Tool to retrieve protein-protein interactions and calculate protein/gene symbol ocurrence in the scientific literature (PubMed & PubMedCentral). Contains two python modules (core and report), and a python script (ppaxe).
Available for python 2.7 and python 3.x, and also as a standalone docker image.
Visit the PPaxe web application to use PPaxe on the web.
S. Castillo-Lara, J.F. Abril
PPaxe: easy extraction of protein occurrence and interactions from the scientific literature
Bioinformatics, AOP November 2018, bty988.
To download and use the ppaxe Docker image:
docker pull compgenlabub/ppaxe:latest
docker run -v /local/path/to/output:/ppaxe/output:rw \
compgenlabub/ppaxe -v -p ./papers.pmids -o ./output.tbl -r ./reportIf you want to install PPaxe manually, go to the Install ppaxe manually section.
usage: ppaxe [-h] -p PMIDS [-d DATABASE] [-o OUTPUT] [-r REPORT] [-i IP] [-v]
[-e]
Command-line tool to retrieve protein-protein interactions from the scientific
literature.
optional arguments:
-h, --help show this help message and exit
-p PMIDS, --pmids PMIDS
Text file with a list of PMids or PMCids
-d DATABASE, --database DATABASE
Download whole articles from database "PMC", or only
abstracts from "PUBMED".
-o OUTPUT, --output OUTPUT
Output file to print the retrieved interactions in
tabular format.
-r REPORT, --report REPORT
Print html report with the specified name.
-i IP, --ip IP Change the IP address of the StanfordCoreNLP server.
Default: http://localhost:9000
-v, --verbose Increase output verbosity.
-e, --exclude Exclude protein symbols not annotated in dictionary.
from ppaxe import core as ppcore
from ppaxe import report
# Perform query to PubMedCentral
pmids = ["28615517","28839427","28831451","28824332","28819371","28819357"]
query = ppcore.PMQuery(ids=pmids, database="PMC")
query.get_articles()
# Retrieve interactions from text
for article in query:
article.extract_interactions()
# Get the predictions
for prediction in article.predictions:
print(prediction.to_html())
# Print html report
# Will create 'report_file.html'
summary = report.ReportSummary(query)
summary.make_report("report_file")# Will read PubMed ids in pmids.txt, predict the interactions
# in their fulltext from PubMedCentral, and print a tabular output
# and an html report
ppaxe -p pmids.txt -d PMC -v -o output.tbl -r report
# Or with docker image
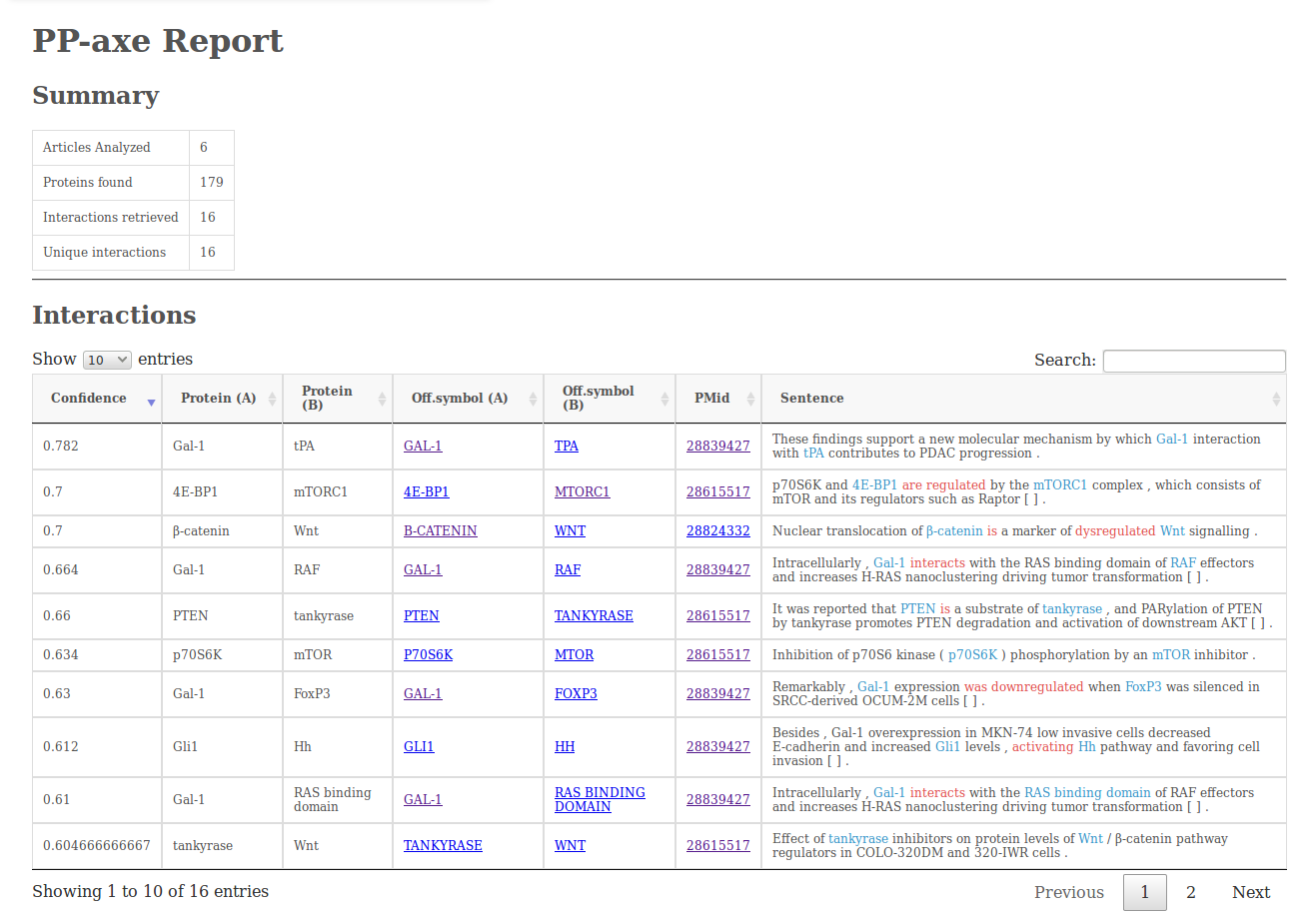
docker run -v /local/path/to/output:/ppaxe/output:rw compgenlabub/ppaxe -v -p pmids.txt -o output.tbl -r reportThe report output (option -r) will contain a simple summary of the analysis, the interactions retrieved (including the sentences from which they were retrieved), a table with the protein/gene counts and a graph visualization made using cytoscape.js.

- Prerequisites
xml.dom
numpy
pycorenlp
cPickle
scipyYou can install this package manuallly using pip. However, before doing so, you have to download the Random Forest predictor and place it in ppaxe/data.
# Clone the repository
git clone https://github.com/scastlara/ppaxe.git
# Download pickle with RF
wget https://www.dropbox.com/s/t6qcl19g536c0zu/RF_scikit.pkl?dl=0 -O ppaxe/ppaxe/data/RF_scikit.pkl
# Install
pip install ppaxe- Download StanfordCoreNLP
In order to use the package you will need a StanfordCoreNLP server setup with the Protein/gene Tagger.
# Download StanfordCoreNLP
wget http://nlp.stanford.edu/software/stanford-corenlp-full-2017-06-09.zip
unzip stanford-corenlp-full-2017-06-09.zip
# Download the Protein tagger
wget https://www.dropbox.com/s/ec3a4ey7s0k6qgy/FINAL-ner-model.AImed%2BMedTag%2BBioInfer.ser.gz?dl=0 -O FINAL-ner-model.AImed+MedTag+BioInfer.ser.gz
# Download English tagger models
wget http://nlp.stanford.edu/software/stanford-english-corenlp-2017-06-09-models.jar -O stanford-corenlp-full-2017-06-09/stanford-english-corenlp-2017-06-09-models.jar
# Change the location of the tagger in ppaxe/data/server.properties if necessary
# ...
# Start the StanfordCoreNLP server
cd stanford-corenlp-full-2017-06-09/
java -mx1000m -cp ./stanford-corenlp-3.8.0.jar:stanford-english-corenlp-2017-06-09-models.jar edu.stanford.nlp.pipeline.StanfordCoreNLPServer -port 9000 -serverProperties ~/ppaxe/ppaxe/data/server.propertiesOnce the server is up and running and ppaxe has been installed, you are good to go.
By default, ppaxe will assume the server is available at localhost:9000. If you want to change the address, set up the server with the appropiate port and change the address in ppaxe by assigning the new address to the variable ppaxe.ppcore.NLP:
- Start the server
# Change the location of the ner tagger in server.properties manually
java -mx10000m -cp ./stanford-corenlp-3.8.0.jar:stanford-english-corenlp-2017-06-09-models.jar edu.stanford.nlp.pipeline.StanfordCoreNLPServer -port your_port -serverProperties ppaxe/data/server.properties- Use the ppaxe package
from ppaxe import core as ppcore
from pycorenlp import StanfordCoreNLP
ppcore.NLP = StanfordCoreNLP(your_new_adress)
# Do whatever you wantBy default, PPaxe uses the HGNC dictionary of gene symbols to normalize the protein/gene symbols found in the article. The ppaxe command-line tool has the option -e that restricts all the results to only those proteins that match against the HGNC database. Users can change this file (located at ppaxe/data/HGNC_gene_dictionary.txt) in order to restrict their searches to only specific genes or proteins, or to normalize gene names using a different dictionary.
Refer to the wiki of the package.
To run the tests:
python -m pytest -v tests
- Sergio Castillo-Lara - at the Computational Genomics Lab
This project is licensed under the GNU GPL3 license - see the LICENSE file for details