
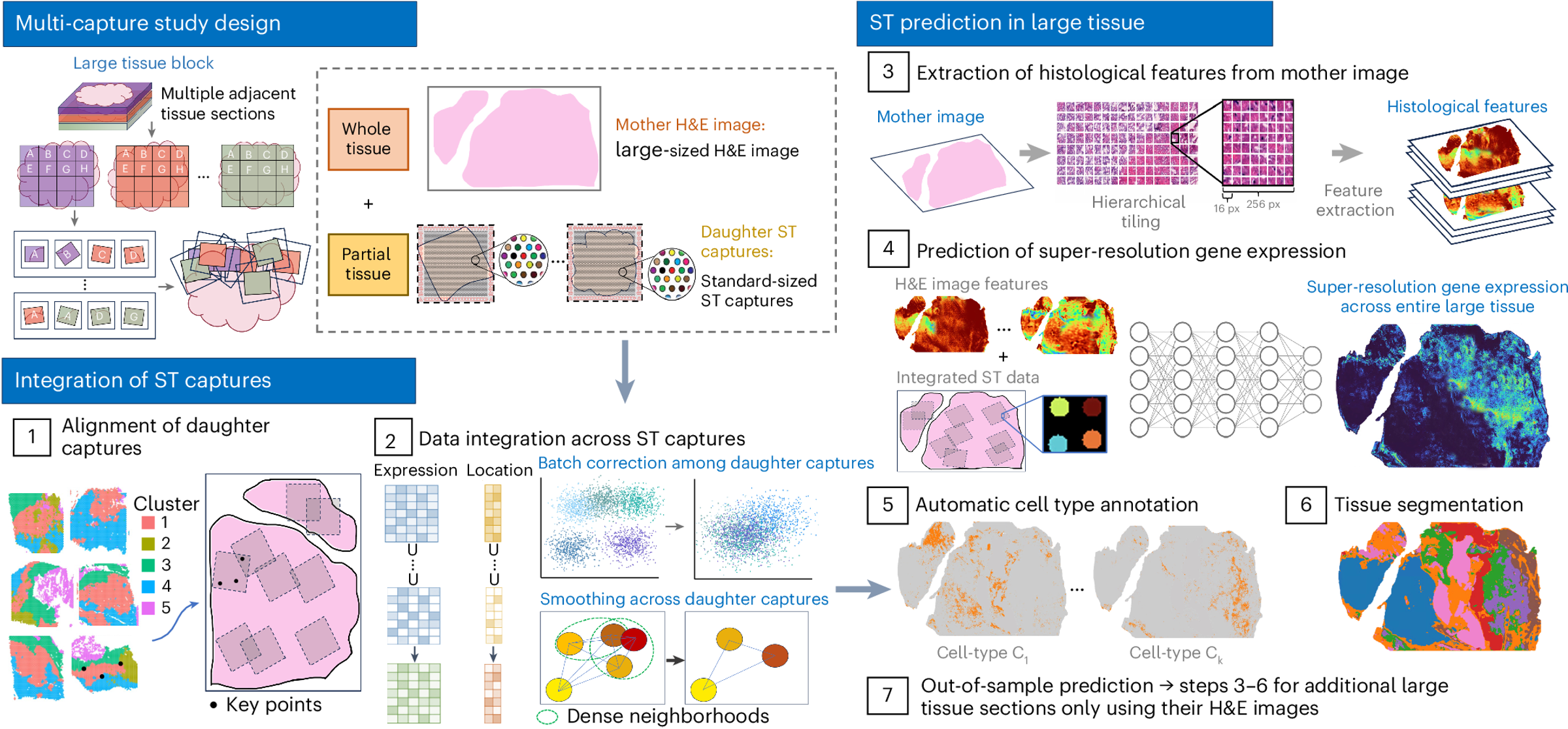
Figure: iSCALE workflow
iSCALE (Inferring Spatially resolved Cellular Architectures for Large-sized tissue Environments) is a novel framework designed to integrate multiple daughter captures and utilize H&E information from large tissue samples, enabling prediction of gene expression with near single-cell resolution across whole-slide tissues.
Clone the repository (recommended) or download the .zip directly from GitHub:
git clone https://github.com/amesch441/iSCALE.git
cd iSCALE-mainconda env create -f environment.yml
conda activate iSCALE_env
cd iSCALEpython -m venv iSCALE_env
source iSCALE_env/bin/activate # Linux/Mac
# or: .\iSCALE_env\Scripts\activate # Windows
pip install -r requirements.txt
cd iSCALE⚡ GPU usage is strongly recommended for speed and scalability. CPU mode is supported but slower.
Download from Box link.
-
Place the model checkpoint files:
vit4k_xs_dino.pthvit256_small_dino.pth
into:
iSCALE-main/iSCALE/checkpoints/ -
Place the
demofolder into:iSCALE-main/iSCALE/data/
To run the demo, submit the appropriate job script depending on your cluster scheduler:
bsub < _run_iSCALE_bsub.sh # For LSF systems
sbatch _run_iSCALE_sbatch.sh # For SLURM systemswith prefix="Data/demo/".
Ground truth for this demo gastric tumor tissue can be found in the cnts-truth-agg folder.
- Use
_run_iSCALE_sbatch.shif your system uses SLURM. - Use
_run_iSCALE_bsub.shif your system uses LSF.
(These scripts are identical except for scheduler setup.)
#SBATCH for SLURM or #BSUB for LSF) to set the correct queue/partition name for your system, as well as any resource requests (GPUs, memory, runtime).
iSCALE-main/
│
├── environment.yml # conda environment specification
├── requirements.txt # pip requirements
│
├── iSCALE/
│ ├── checkpoints/ # pretrained models (place downloaded .pth files here)
│ ├── data/ # input data (demo folder goes here)
│ ├── Alignment_scripts/ # tools for semi-automatic alignment
│ ├── logs/ # log directory
│ │ ├── logs_output/ # job standard output logs
│ │ └── logs_errors/ # job error logs
│ ├── *.py # main Python scripts
│ ├── *.sh # run scripts (SLURM/LSF)
│ └── ...
Each project has the following structure:
iSCALE-main/iSCALE/Data/<project_name>/
│
├── DaughterCaptures/
│ ├── UnallignedToMother/ # raw ST data (Visium, Visium HD, Xenium, CosMx)
│ │ ├── D1/
│ │ │ ├── cnts.tsv # count matrix (genes × spots)
│ │ │ ├── locs.tsv # coordinates (spot_id, x, y)
│ │ │ └── he.* # H&E image (see formats below)
│ │ ├── D2/
│ │ └── ...
│ │
│ └── AllignedToMother/ # aligned data (produced after registration)
│ ├── D1/
│ │ ├── cnts.tsv
│ │ └── locs.tsv
│ ├── D2/
│ └── ...
│
└── MotherImage/
├── he-raw.* # raw H&E (before scaling)
├── he-scaled.* # scaled H&E (after resizing)
├── he.tiff # final processed H&E with padding
├── radius-raw.txt # raw spot radius in pixels
├── radius.txt # scaled radius in pixels (auto-generated if missing using rescale_locs.py)
└── markers.csv (optional) # marker genes for auto-annotation
- Always run
preprocess.pyto generate the finalhe.tifffile for the MotherImage folder. - Supported input H&E formats for mother image:
.tiff,.tif,.svs,.ome.tif,.ome.tiff,.jpg,.png,.ndpi,.scn,.mrxs - locs.tsv: must contain
spot x y - cnts.tsv: genes × spots matrix (tab-delimited).
- markers.csv (optional):
gene,label MKI67,Tumor KRT20,Mucosa ...
Parameters are set in the run scripts (_run_iSCALE_sbatch.sh or _run_iSCALE_bsub.sh).
| Parameter | Description | Default Example |
|---|---|---|
prefix_general |
Project directory path (must contain DaughterCaptures and MotherImage) |
Data/demo/ |
daughterCapture_folders |
List of daughter capture folders | ("D1" "D2" "D3" "D4" D5") |
device |
Compute device: "cuda" (GPU) or "cpu" |
"cuda" |
pixel_size_raw |
Pixel size (µm/pixel) of raw H&E | 0.252 |
pixel_size |
Desired pixel size after rescaling | 0.5 |
n_genes |
Number of most variable genes to impute | 100 |
n_clusters |
Number of clusters for downstream analysis | 20 |
dist_ST |
Smoothing parameter across ST captures (integration sharpness) | 100 |
Notes
prefix_generalis the main project folder.dist_ST=100works well in most cases, but check QC plots iniSCALE_output/spot_level_st_plots/spots-integratedto tune if needed.n_genes=100is used in the demo because the Xenium dataset has a small targeted panel. For Visium and other platforms with larger gene counts, much higher values (e.g. 3000) are appropriate.
All results are saved to iSCALE_output/:
- spot_level_st_plots/
QC plots to confirm correct alignment of daughter captures onto mother image. - super_res_gene_expression/
Imputed super-resolution expression (pickle files).refined/subfolder updates predictions for regions unlikely to contain cells.
- super_res_ST_plots/
Visualizations of super-resolution gene expression.- includes
refined/.
- includes
- clusters-gene_#/
Clustering results using imputed gene expression. - annotation/
Cell-type/region annotations if markers.csv was provided.
If you use iSCALE, please cite:
Schroeder A., et al. Scaling up spatial transcriptomics for large-sized tissues: uncovering cellular-level tissue architecture beyond conventional platforms.
Nature Methods (2025).
https://www.nature.com/articles/s41592-025-02770-8
This project is licensed under the terms of the LICENSE file included in this repository.