
A computational biology toolkit for Mars terraforming research, combining molecular docking, metabolic modeling, and genetic engineering workflows tailored for Mars environmental conditions.
Static site hosted on Vercel: https://reddust-reclaimer.vercel.app
This is a fully static HTML export of the Sphinx documentation located in docs/dist/.
No Python runtime is required for the deployed version — you only need Python locally if building docs or running the toolkit.
- Python 3.8–3.11
- Git
- Optional: Micromamba/Conda (recommended for reproducible environments)
- Optional: Docker
git clone https://github.com/Jonahnki/reddust-reclaimer.git
cd reddust-reclaimer
micromamba create -y -f environment.yml
micromamba activate reddust-reclaimer
python -m pip install -e . pytest-xdistRun example workflows:
python -m reddust_reclaimer.dock_example --help
python -m reddust_reclaimer.codon_optimization --sequence ATGCGATCGTAGC --analyze
python -m reddust_reclaimer.metabolic_flux --model models/mars_microbe_core.xmlFor maintainers, deploying docs to Vercel:
./docs/build.sh
git add -f docs/dist
git commit -m "Build: update static docs"
git pull --rebase origin main
git push origin mainVercel is configured via vercel.json to:
- Skip Python installation
- Serve
docs/distas the site root
- Mars-adapted enzyme docking simulating protein–ligand interactions under Mars conditions
- Atmospheric processing with emphasis on CO2 fixation and extremophile pathways
- Accounts for low temperature, high radiation, and low pressure
- Sequence optimization tailored for Mars extremophile adaptation
- Codon usage stability across −80°C to 20°C
- Emphasis on radiation-resilient encoding choices
- Flux balance analysis (FBA) for efficient Mars resource utilization
- Modeling of CO2 fixation pathways informed by Mars atmospheric data
- Optimization for water and energy use under scarcity
from reddust_reclaimer.dock_example import MarsEnzymeDocking
# Initialize for Mars conditions (T in Kelvin, P in bar)
docker = MarsEnzymeDocking(temperature=233.15, pressure=0.006)
# Run docking simulation
results = docker.dock_mars_enzyme_substrate("carbonic_anhydrase")
docker.print_docking_summary(results)from reddust_reclaimer.codon_optimization import MarsCodonOptimizer
optimizer = MarsCodonOptimizer()
optimized_seq = optimizer.optimize_for_mars_conditions("ATGAAATTTGGGTAG")
print(f"Optimized: {optimized_seq}")from reddust_reclaimer.metabolic_flux import MarsMetabolicNetwork
network = MarsMetabolicNetwork()
results = network.mars_metabolic_flux_analysis("biomass_synthesis")
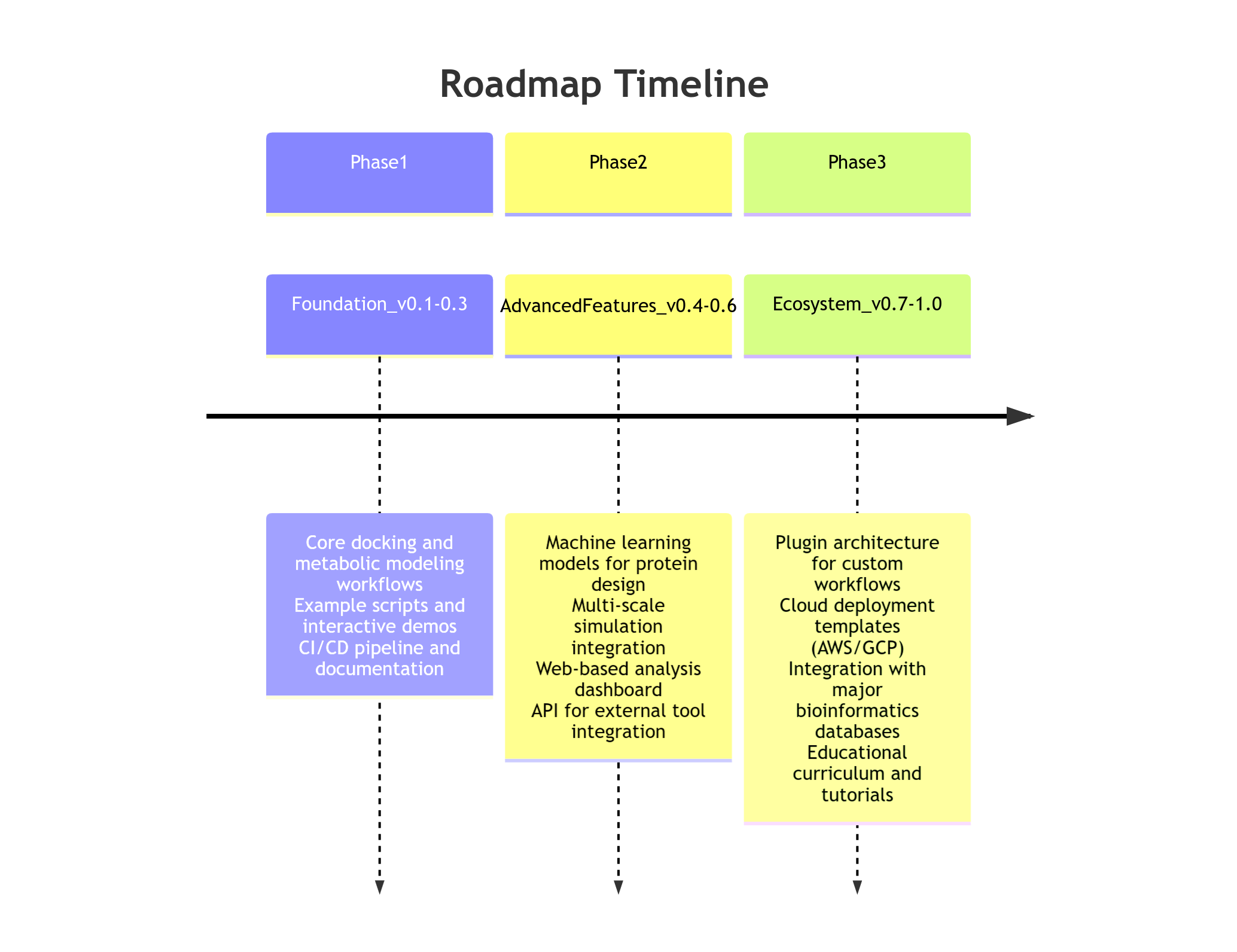
network.print_flux_analysis(results)- Core docking and metabolic modeling workflows
- Example scripts and interactive demos
- CI/CD pipeline and documentation
- Machine learning models for protein design
- Multi-scale simulation integration
- Web-based analysis dashboard
- API for external tool integration
- Plugin architecture for custom workflows
- Cloud deployment templates (AWS/GCP)
- Integration with major bioinformatics databases
- Educational curriculum and tutorials
Run the full test suite:
pytest tests/ -v --cov=reddust_reclaimerTest individual components:
# Docking workflow
python -m reddust_reclaimer.dock_example --verbose
# Codon optimization
python -m reddust_reclaimer.codon_optimization --sequence ATGAAATTTGGGTAG --analyze
# Metabolic flux analysis
python -m reddust_reclaimer.metabolic_flux --plotreddust-reclaimer/
├── reddust_reclaimer/ # Package: workflows and modules
│ ├── dock_example.py # Protein–ligand docking demo
│ ├── codon_optimization.py # Genetic sequence optimization
│ └── metabolic_flux.py # Flux balance analysis
├── models/ # Biological models and data
│ ├── mars_microbe_core.xml # SBML metabolic model
│ ├── protein_structures/ # Sample PDB files
│ └── compound_library.sdf # Mars-relevant compounds
├── data/ # Sample datasets
├── notebooks/ # Interactive Jupyter demos
├── tests/ # Test suite
└── .github/ # CI/CD and community templates
Contributions are welcome! Please review the Contributing Guidelines and Code of Conduct.
git clone https://github.com/Jonahnki/reddust-reclaimer.git
cd reddust-reclaimer
pip install -r requirements.txt
pip install -e .
# Run tests
pytest tests/
# Code formatting and linting
black reddust_reclaimer/ tests/
flake8 reddust_reclaimer/ tests/pre-commit install # Enable git hooks
pre-commit run --all-files # Run hooks on all filesmake install # Install dependencies and package
make test # Run tests with coverage
make format # Apply black formatting
make format-check # Check formatting compliance
make lint # Run flake8 + mypy
make examples # Run example scripts
make docs # Build Sphinx documentationA minimal Nextflow workflow is provided at workflows/nextflow/main.nf. Run with conda environment:
nextflow run workflows/nextflow/main.nf \
--sequence ATGAAATTTGGGTAG \
--model models/mars_microbe_core.xml \
--outdir results/nextflow \
--threads 2Parameters:
--sequenceCodon optimization input sequence (default: example sequence)--modelSBML metabolic model (default: models/mars_microbe_core.xml)--outdirOutput directory (default: results/nextflow)--threadsNumber of CPU threads (default: 2)--dry_runUse--dry_run trueto test pipeline without heavy computation
The default Nextflow config (workflows/nextflow/nextflow.config) enables conda and sensible defaults. A Docker profile is available (-profile docker) if an image is built/published.
If you use this toolkit in your research, please cite:
@software{reddust_reclaimer,
title = {RedDust Reclaimer: Computational Biology Toolkit for Mars Terraforming},
author = {John Adedeji},
year = {2025},
url = {https://github.com/Jonahnki/reddust-reclaimer},
version = {0.1.0}
}This project is licensed under the MIT License — see the LICENSE file for details.
- Mars atmospheric data from NASA JPL
- Extremophile genomic data from NCBI
- Metabolic modeling frameworks: COBRApy, SBML
- Molecular docking tools: RDKit, AutoDock Vina
- CI powered by GitHub Actions using Micromamba and Python 3.8–3.11 matrix (
.github/workflows/ci-conda.yml) — all tests passing ✅ - Linting (flake8/black), typing (mypy), tests (pytest + coverage), notebook execution, security scans (pip-audit, bandit), Docker image builds
- Artifacts include coverage reports, executed notebooks, processed data, documentation builds, results, and logs
- Legacy pip-based CI (
.github/workflows/ci.yml) is retained for manual runs only
-
RDKit installation issues:
- On Linux, if
rdkit-pypifails, update pip/setuptools/wheel:python -m pip install --upgrade pip setuptools wheel - Prefer conda-forge package:
conda install -c conda-forge rdkitand remove piprdkit-pypidependency
- On Linux, if
-
libSBML errors (ImportError: libsbml not found):
- Install via conda:
conda install -c conda-forge python-libsbml(included inenvironment.yml) - For pip-only, ensure system libs installed; consider switching to a conda environment
- Install via conda:
-
macOS M1/M2/M3 or ARM runners:
- Use conda-forge packages; some pip wheels may be unavailable on ARM
- Docker fallback recommended
-
Pre-commit fails on notebooks:
nbQAruns Black and Flake8 over notebooks- To skip files, add excludes to
.pre-commit-config.yamlor commit with-nto bypass temporarily