F1score
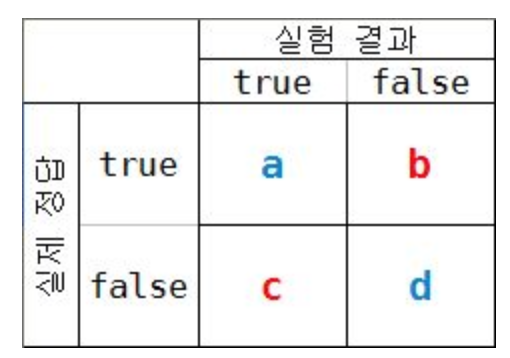
- TT : 실제 정답 T, 실험 결과 T (a)
- TF : 실제 정답 T, 실험 결과 F (b)
- FT : 실제 정답 F, 실험 결과 T (c)
- FF : 실제 정답 F, 실험 결과 F (d)
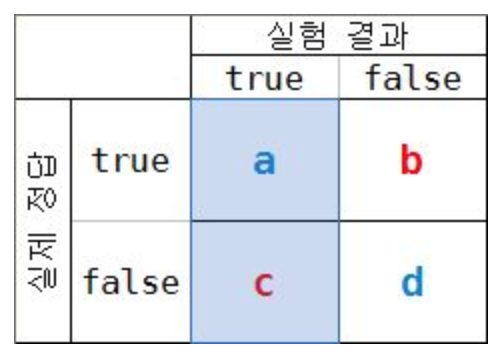
- a/(a+c)
- 실험 결과 True라고 판단된 것 중에 실제 정답이 True인 것
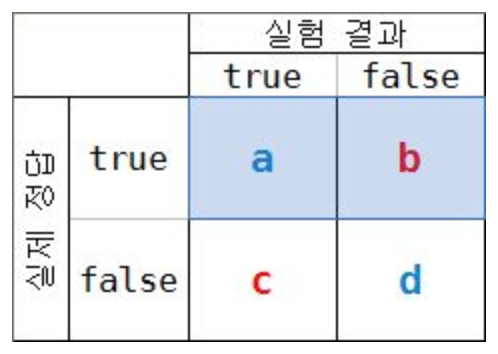
- a/(a+b)
- 실제 정답이 True인 것 중에 실험 결과가 True인 것
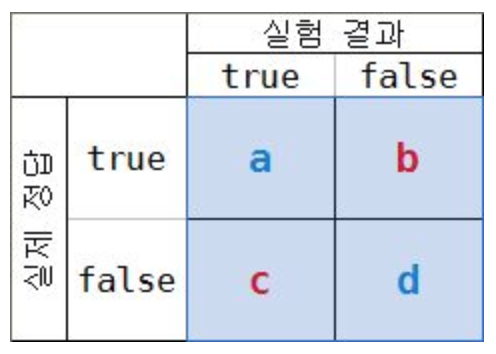
- (a+d)/(a+b+c+d)
- 전체 결과 중에서 정답인 것
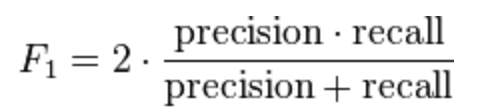
- precision과 recall에 대한 평균에 가중치를 주는 것

- macro average
- 클래스 별 f1 score에 가중치를 주지 않는다.
- 클래스의 크기에 상관 없이 모든 클래스를 같은 비중으로 다룬다
-

- (학교의 각 반 성적)
- micro average
- 모든 클래스의 FP, FN, TP, TN의 총 수를 센 후 precision, recall, f1 score를 수치로 계산
- 전체적인 성능을 나타낸다
-

- (전체 학생들의 성적)
- 각 샘플을 똑같이 간주한다면 micro average, 각 클래스를 동일한 비중으로 고려하면 macro average 사
- 두 문자열의 유사도를 판단
- 문자열 A를 B로 바꾸기 위해 필요한 연산의 최소 횟수
- 비교할 두 문자가 같으면 cost(i,j) = cost(i-1, j-1)
- 비교할 두 문자가 다르면 cost(i,j) = 1 + min( cost(i-1,j),cost(i,j-1),cost(i-1,j-1) )
def _edit_dist_init(len1, len2):
A = []
for i in range(len1):
A.append([0] * len2)
# (i,0), (0,j) 채우기
for i in range(len1):
A[i][0] = i
for j in range(len2):
A[0][j] = j
return A
def _edit_dist_step(A, i, j, s1, s2, transpositions=False):
c1 = s1[i-1]
c2 = s2[j-1]
a = A[i-1][j] + 1 # s1에서 skip
b = A[i][j-1] + 1 # s2에서 skip
c = A[i-1][j-1] + (c1!=c2) # 대체
d = c+1 # X select
if transpositions and i>1 and j>1:
if s1[i-2] == c2 and s2[j-2] == c1:
d = A[j-2][j-2] + 1
A[i][j] = min(a,b,c,d)
def edit_distance(s1, s2, transpositions=False):
len1 = len(s1)
len2 = len(s2)
lev = _edit_dist_init(len1 + 1, len2 + 1)
for i in range(len1):
for j in range(len2):
_edit_dist_step(lev, i+1, j+1, s1, s2, transpositions=transpositions)
return lev[len1][len2]
- 두 개의 객체를 집합으로 간주하여 유사성을 측정
def jacc_sim(query, document):
a = set(query).intersection(set(document))
b = set(query).union(set(document))
return len(a)/len(b)
- 보통 DNA 서열 검출을 위해 사용